Understanding the market dynamics of biosimilars
Abstract
Biosimilars represent an attractive market opportunity in the pharmaceutical industry. In order to understand the underlying market dynamics and to identify the success factors of the biosimilars market, a PEST (political, economic, social, technological) analysis was conducted based on desk research and expert interviews with market participants and stakeholders. The regulatory environment for biosimilars seems to be well established and both the required manufacturing technology and the necessary analytical capabilities for biosimilar development are available. The potential market is expected to grow due to the overall dynamics in the biologics market and the patent expiration of blockbuster drugs. The perspective of the scientific community towards biosimilars has changed from skeptical to rather positive in the last 10 years, probably reflecting the evolution of regulatory guidelines and technological progress. However, physicians, responsible for the prescription of drugs, are still rather skeptical about biosimilars and need to be better informed in order to increase the currently low market penetration of biosimilars. Taken together, the biosimilars industry is expected to step out of its infancy stage and now enter the growth phase.
1 Introduction
In 1982, the company Eli Lilly received approval for the first recombinant protein, i.e. human insulin, for therapeutic use in the EU. More than 30 years later, in 2015, a total of 185 biotechnologically-derived drugs has been approved (VFA, 2015). They are applied in various therapeutic areas, such as oncology (e.g., monoclonal antibodies against breast cancer), haematology (e.g., erythropoietin or epoetin against anemia), bleeding disorder (e.g., Factor VIII) or immunology (e.g., interferon alfa).
These biotechnologically-derived drugs are called “biopharmaceuticals” or “biologics”. The latter term will be used in this article, following the nomenclature of de Mora (2015). As shown in table 1, biologics differ from medicinal products that are chemically synthesized such as acetylsalicylic acid, which will further be referred to as “chemically-synthesized drugs”, in a number of ways (Dingermann and Zündorf, 2014; ProGenerika, 2014a, Schellekens, 2004).
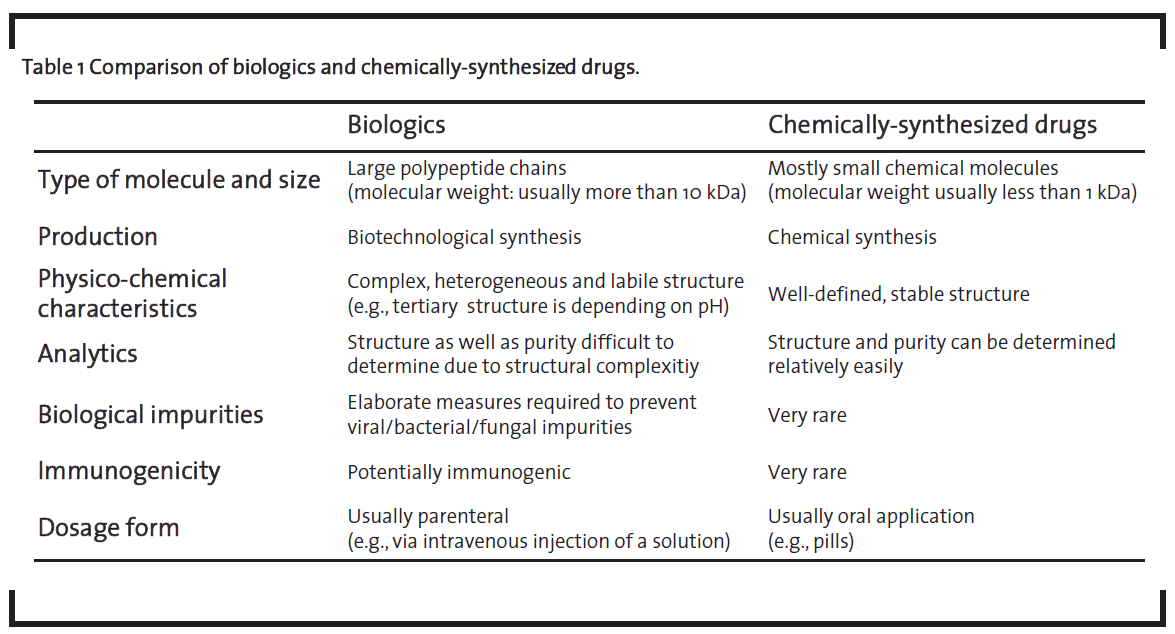
Biologics are significantly larger than chemically-synthesized drugs. Their size can be up to several hundred kDa in the case of monoclonal antibodies. Due to their size, they cannot be manufactured by chemical synthesis but have to be produced by using biotechnological processes in living cells. Biologics are complex molecules with therapeutic efficiency depending on their higher-order structure, especially the tertiary structure of the protein, and the pattern of post-translational modifications such as glycosylation or disulfide bridges. Thus, biologics have to be administered parenterally as they otherwise would undergo significant structural modifications in the gastrointestinal tract. For example, a protonation of basic amino acid side chains in the acidic environment in the stomach would lead to the denaturation of the protein and thus nullifying the desired therapeutic effect. The inherent structural complexity of biologics combined with the high quality level required for intravenous administration, is one of the key differences of this class of medicinal products compared to chemically-synthesized drugs. This is reflected by an elaborate and expensive development process as well as the high prices of biologics, which are in turn increasing therapy costs. For example, the price for five doses of Remicade® is reported to amount to EUR 4,675 (Apotheke adhoc, 2015).
As the patents of originator products, i.e. the biologics that have first entered the market, expired or are about to expire, the market is now open for generic producers to enter this seemingly attractive market. Generic manufacturers usually produce a molecular copy of the originator product and sell it at a lower price. This is also very attractive for health insurance companies in order to reduce the financial burden on the healthcare system.
However, it is not possible to produce an exact copy of the original molecule. Even minor differences in the manufacturing process will e.g. result in a different glycosylation pattern of the product and thus yield in a different molecule. The European Medical Agency (EMA) thus developed the expression “biosimilar” in their 2005 guideline (EMA, 2005). A biosimilar is a medicinal product shown to be essentially the same as the original product but not identical. The term “biogeneric” is therefore not suitable for biologics and should not be applied. In the context of this article, it is further important to distinguish between first-wave and second-wave biosimilars (Hinderer, 2012; Schellekens, 2015). The first refers to smaller and “simple” biomolecules such as insulin or the growth hormone somatropin, which are produced in E. coli or yeast cells and usually do not show post-translational modifications. The second term refers to larger biomolecules such as monoclonal antibodies that exhibit a much broader structural diversity and which are usually produced in mammalian cells giving way to different post-translational modifications.
In this work, the status quo of the market for biosimilars is analyzed along the structural framework of a PEST analysis (P: political environment, E: economic environment, S: social environment, T: technological environment) with primary focus on the following questions:
- Political environment: How far developed and reliable is the regulatory framework for the introduction of biosimilars? What is required to obtain marketing authorization for a biosimilar? Are there companies that have successfully run through the authorization process in the EU?
- Economic environment: How large is the market of biologics that have already lost or are losing patent protection? How will the competitive environment in the biosimilars market evolve – and will there be a difference to the “classical” chemically-synthesized generics?
- Social environment: What is the role of key stakeholders such as health insurance companies, physicians or patients in the biosimilars market? What is their attitude towards biosimilars and to what extent do they affect market dynamics?
- Technological environment: What are the key competencies required in biosimilars development, especially with respect to project management? To what extent are analytical techniques (already) capable to demonstrate “similarity” between a biosimilar and its reference product?
Based on this status quo, an outlook on the biosimilars market is established and management implications for different types of market players are discussed.
2 Methods
2.1 PEST analysis
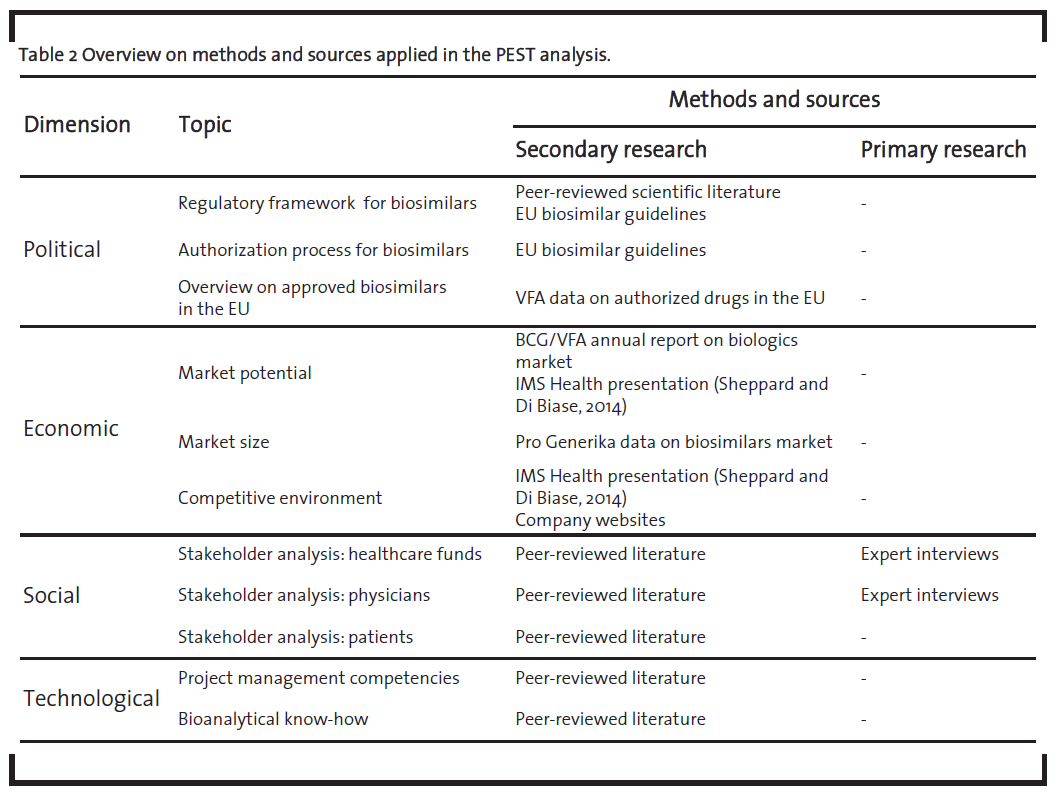
The PEST analysis was mainly based on secondary research supplemented with primary research where applicable and appropriate. Table 2 provides an overview on the sources used.
2.2 Market outlook
In order to monitor the market dynamics of the last years and to establish a sound database for a market outlook, 234 peer-reviewed scientific articles and conference contributions (referred to as “sources” in the following) containing the keyword “biosimilar” were analyzed. Those sources were selected by conducting a search for the keyword “biosimilar*” either in the title or abstract of a publication within the timeframe from 2004 to 2015 in the scientific database Sciencedirect (Sciencedirect, 2015). Each of the sources was classified in one of the three categories “skeptical”, “neutral” and “positive”, depending on its characteristic content. The source was classified as “sceptical” when the text rather questions the viability of biosimilars and discusses mainly problems in the context of biosimilars. A text is classified as “neutral” when it does not generally question the viability of biosimilars and discusses both problems and suggestions for solutions. The source was classified as “positive” when the text acknowledges the potential of biosimilars and discusses solutions rather than highlighting problems.
3 Results
3.1 PEST analysis
3.1.1 Political perspective
The EU was the first region to establish regulatory guidelines for the authorization of biosimilars (EMA, 2005). These guidelines reflect the complexity of biologics on a regulatory level as it requires a substantial amount of pre-clinical and clinical studies before obtaining marketing authorization (figure 1). While pre-clinical studies are required in order to confirm similarity on a molecular level, clinical studies are required to confirm the therapeutic efficiency of the biosimilar.
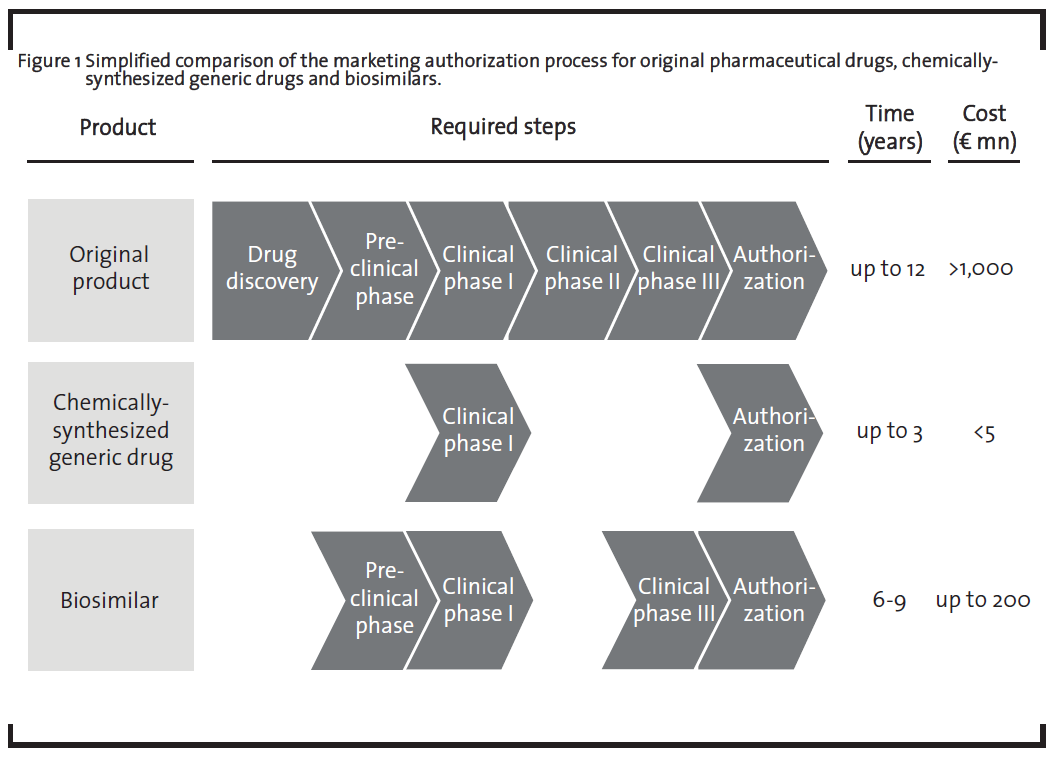
The necessity of the clinical phase III in the development of biosimilars is required due to the fact that structural differences such as a different glycosylation pattern in the biosimilar compared to the original drug may affect the therapeutic efficiency of the medicine. Companies planning to market a biosimilar thus need to demonstrate that their product is at least as effective as the original drug. It is possible that once the effectivity of the biosimilar is established for one indication, the results can be extrapolated to other indications. For example, the biosimilar monoclonal antibody infliximab was approved in the EU in 2013 for all indications of the originator product (Remicade®, Johnson & Johnson), including rheumatoid arthritis, psoriasis arthritis, psoriasis, inflammatory bowel diseases and ankylosing spondylitis (GaBiOnline, 2013). However, in Canada, the same biosimilar was only approved for autoimmune arthritis as an extrapolation of the clinical data to inflammatory bowel diseases was not granted (GaBiOnline, 2015a). Extrapolation to other indications thus remains a subject of debate, which is also underlined by the discussion in the scientific community (Weise, 2014). The necessity of the pre-clinical phase as well as the clinical phase III in the biosimilar development marks the major difference to chemically-synthesized generic drugs which are only required to demonstrate their bioequivalence with the original drug in clinical phase I. A comprehensive discussion on the preclinical development of biosimilars can be found in a recently published paper by Bui et al. (2015). Additionally, after having obtained marketing authorization, companies further need to implement a pharmacovigilance plan, i.e. further studies in order to rule out potential long-term negative side effects.
This different extent in pre-clinical and clinical studies leads to significant higher development costs for biosimilars which may reach up to EUR 200 mn, representing 20% of the development costs of the original drug (ProGenerika, 2014b), compared to the development of chemically-synthesized generic drugs which require less than 1% of the development costs of the original drug (Mellstedt, 2013). This also results in a lower price reduction compared to the original drug. Prices for biosimilars are usually set 10 to 30% below the originator price (Tsuruta et al., 2015), but there is one example of a biosimilar that was introduced at a 72% discount in Norway (GaBiOnline, 2015b). The market price for chemically-synthesized generic drugs are usually more than 80% below the price of the original product. On the other hand, both chemically-synthesized generic drugs and biosimilars do not need to run through the “drug discovery” phase as the therapeutic target of the medicinal product is already known. The clinical phase II is also not required as the dosage of the biosimilar is the same as for the original product.
As shown in table 3, a total of 18 biosimilars received marketing authorization from the EMA between 2006 and 2014 (VFA, 2015). This underlines the applicability of the EMA guidelines.
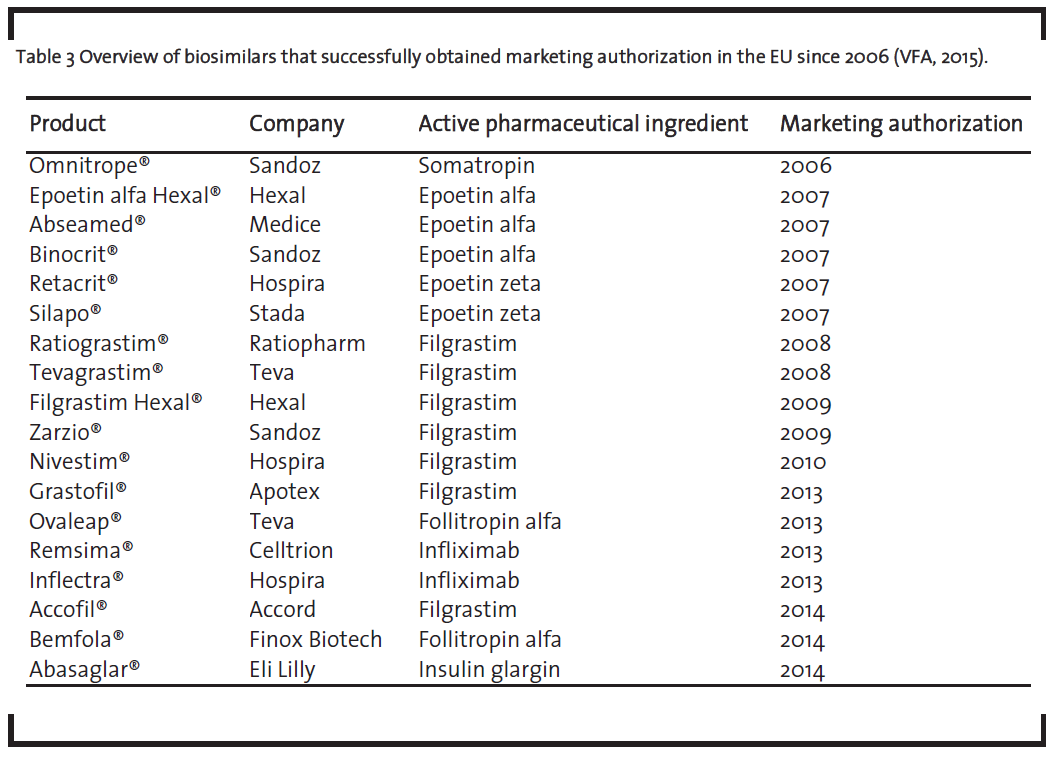
One should note that the list of biosimilars in table 3 also contains the monoclonal antibody infliximab, which received approval. This is remarkable as monoclonal antibodies represent the most complex biologics and their development as biosimilar was seen very skeptical some years ago (Schneider and Kalinke, 2008). Given the fact that the development of biosimilar monoclonal antibodies is nevertheless possible, it underlines the broad applicability of the EMA framework.
After the establishment of biosimilar guidelines in the EU, more than 30 countries worldwide have also set up their own guidelines (Heinemann et al., 2015). These countries comprise large pharmaceutical markets such as Japan, where guidelines were established in 2008, for Canada in 2009, for the US in 2010 and for India in 2011, respectively. In China, regulatory guidelines are currently in the evolution process (Heinemann et al., 2015). This evolving regulatory environment for biosimilars will continue to give companies a reliable framework for their development process.
3.1.2 Economic perspective
The market potential for biosimilars is primarily driven by the market growth of biologics which is in turn determined both by the growing number of biologics on the market as well as their high price. The market of biologics has grown significantly in recent years (figure 2). In 2006, biopharmaceuticals already represented 12% of the German pharmaceuticals market value – by 2014, the market share of biopharmaceuticals had raised to 22%. During this period, biopharmaceuticals grew with a compound annual growth rate (CAGR) of 12%, whereas chemically-synthesized drugs rather stagnated with a 1% CAGR as shown in figure 2 (BCG, 2006-2015).
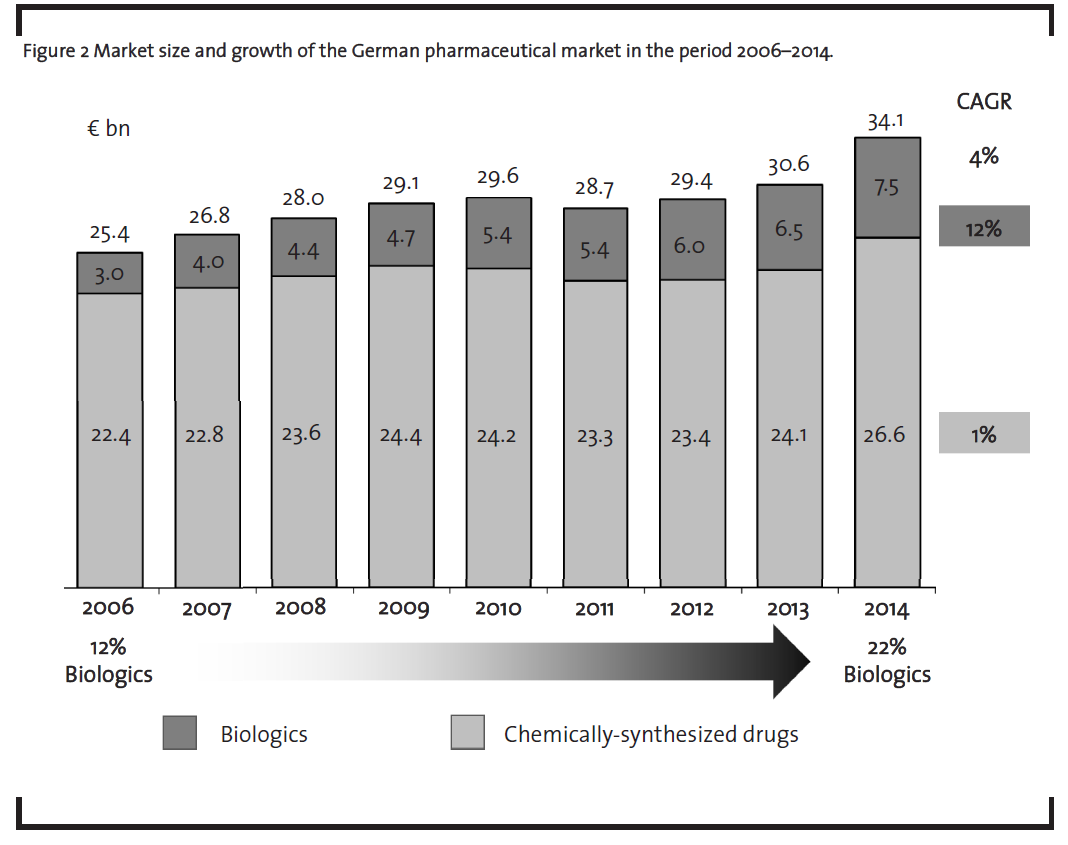
Furthermore, more than ten blockbuster biologics, i.e. drugs with annual sales of more than EUR 1bn, will lose their patent protection in the period from 2013 to 2019 (Sheppard and Di Biase, 2014) as demonstrated in table 4.
This underlines the market attractiveness from a sales perspective. As shown in section 3.1.1, companies already start to enter this market. However, as can be seen from figure 3, biosimilars demonstrate only a limited market penetration so far (Pro-Generika, 2015).
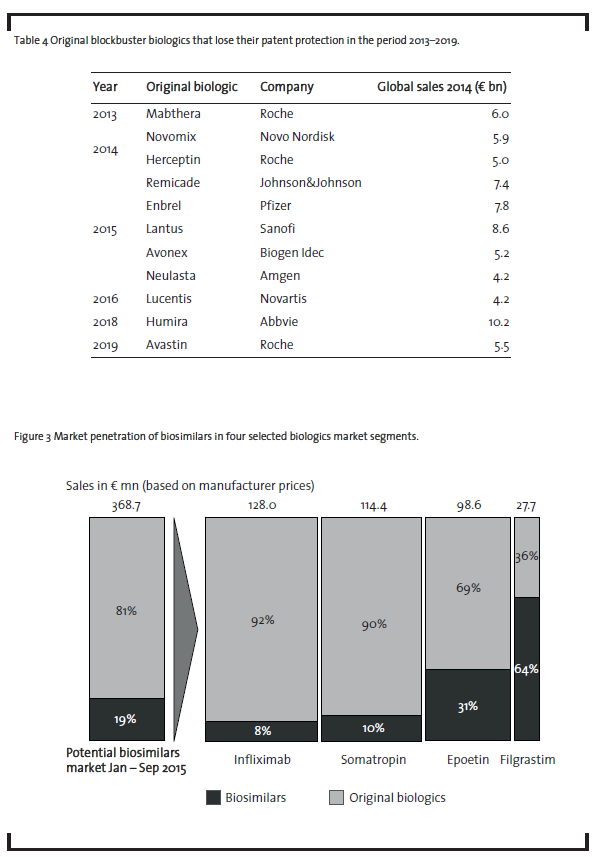
Currently, biosimilars containing infliximab, somatropin, epoetin or filgrastim only represent less than one fifth of the German market for biologics that have already lost patent protection. However, there are significant differences in the market penetration depending on the product. With 8% market penetration, the market for the monoclonal antibody infliximab currently shows the lowest values.However,this biosimilar has only been introduced on the market in February 2015 and reached about 18% market share in October 2015 (ProGenerika, 2015). It remains to be seen whetherthis share can be increased in the upcoming months. In the case of somatropin, only one biosimilar player, i.e. Sandoz, is competing against an oligopoly of six companies producing the original product: Pfizer, Novo Nordisk, Merck, Eli Lilly, Ferring and Ipsen Pharma. This competitive environment makes market penetration forthe biosimilar drug difficultto achieve. Competition based on price alone will be difficult due to the low price reduction of the biosimilar compared to the original product as discussed above. Epoetin biosimilars have achieved a somewhatlarger market penetration, mainly due to the fact that epoetin therapies are mostly short-term therapies which makes it easier to switch from the original product to a biosimilar. In this context, it is important to note that a biosimilaris usually applied from the beginning of a therapy and physicians are reluctant to substitute an original biologic during an ongoing therapy. Filgrastim biosimilars have achieved the largest market penetration, probably due to the fact that seven biosimilar players have received marketing authorization (see table 3),whereas there is only one company on the market, Amgen, who markets the original product. However, given the relatively small size of the filgrastim market, this does not change the overall picture of a currently low market penetration of biosimilars.
The competitive environment in the biosimilars market is shaped by at least three different types of companies:
- Traditional generics manufacturer (e.g., Teva, Sandoz, Stada),
- Large pharmaceutical companies (e.g.,Eli Lilly),
- New market entrants (e.g., Celltrion).
Traditional generics companies were the main driving force behind the first wave of biosimilars, such as somatropin, filgrastim and epoetin. Their development is less complex (and probably less cost intensive) compared to second-wave biosimilars, so that the entrance of some traditional generics player is not surprising. However, so far there have only been attempts but no classical generics company has successfully developed a second-wave biosimilar, which require more initial investments and bear a higher risk. The company Stada invested in a development program for the biosimilar trastuzumab, a monoclonal antibody, but canceled the project in 2010 and further decided to follow an in-licensing strategy (Stada, 2011). The company Teva, together in a joint venture with the Swiss company Lonza, stopped clinical trials on their biosimilar rituximab in 2012 (Biosimilar News, 2012).
Biosimilars also present an (additional) attractive market for large pharmaceutical companies (Ledford, 2010). As the price of a biosimilar is usually at least 70% of the originator price, it is attractive for these companies to invest in development programs of biosimilars as well. One significant advantage compared to the development of novel pharmaceutical drugs is the fact that the pharmaceutical target is already known, which means that there is no need for cost-intensive research and development in the early phase of drug development and a higher probability of success. Additionally, large pharmaceutical companies benefit from their development expertise and financial strength to approach the development of second-wave biosimilars. Indeed, looking at the development pipeline of biosimilar monoclonal antibodies, one can observe that large pharmaceutical companies such as Boehringer Ingelheim, Pfizer and Merck are currently trying to enter this market (ProGenerika, 2014b).
Another strategy for established large biopharmaceutical companies like Amgen or Sanofi is the development of so-called “biobetters”, i.e.improved versions of the original product that again receive patent protection (Casey, 2015). Examples would be insulin glargin (e.g., Sanofi’s human insulin analogue Lantus®) or a PEGylated form of filgrastim (e.g., Amgen’s Neulasta®). The development of a biobetter can be interpreted as a strategy of original producers pursuing patent extension and will not be discussed in detail here as it was discussed elsewhere (Casey, 2015).
Most interestingly, there are also new market entrants that are stirring up the competitive environment. One of the most intriguing case examples is the company Celltrion (South Korea). Founded in 2002, the company received EU approval for the monoclonal antibody infliximab (product: Remsima) in 2013 and started to enter the market in 2015 (after patent expiration of the original product). Celltrion can be classified as a so-called “emerging market player”, a company thatfirstfocuses on a certain number of regional markets (e.g., India) before attempting to enter the highly regulated pharmaceutical markets in the EU and the USA (Sheppard and Di Biase, 2014).
3.1.3 Social environment
In order to evaluate the social environment, three different stakeholders involved in the market acceptance of biosimilars are examined:
- Health insurance companies as they are responsible for refinancing the therapies,
- Physicians as they decide aboutthe application of a biosimilar versus the original biologic,
- Patients as they are in the end the market participantswho receive treatment with the biosimilar.
Pharmacies were not included in the analysis as they are not involved in the decision process about whether a biosimilar or an original drug should be used. This is a sharp contrast to chemically-synthesized generics, where automatic substitution at the pharmacy gives the pharmacist a completely different role.
Health insurance companies are generally open towards biosimilars as they represent a means to reduce the financial burden on healthcare systems. Even if the price reduction is usually only in the order of 10 to 30% compared to the original biologic drug, this still represents huge potential savings for the healthcare system. In order to create incentives for physicians to favor biosimilars during the prescription of drugs, healthcare funds introduced target agreements in some German regions (ProGenerika, 2014a). For example, the Association of Statutory Health Insurance Physicians (Kassenärztliche Vereinigung) of the federal state Bremen included specific prescription target quotas in its annual agreement with the healthcare insurance companies (KV Bremen, 2015). As a result, Bremen belonged to the federal states in Germany with the highest prescription quota for biosimilars (ProGenerika, 2015).
The typical discount contracts that are usually applied after patent expiration of a drug are not seen as an instrument to improve the usage for biosimilars by healthcare insurance companies. Given the high development costs for biosimilars, discount contracts rather favor the original producer who already had the opportunity to earn their return on investment. For a biosimilar producer, discount contracts would represent a higher risk as they would be trapped between earning their investment and realizing a low price to be attractive for health insurance companies.
Physicians are the most important stakeholder group as they are responsible for prescribing the biosimilars. However, they are currently rather skeptical towards biosimilars as they are not sufficiently informed. In particular, they are concerned about the pharmaceutical quality of biosimilars, their safety (immunogenicity potential), efficacy (extrapolation in clinical studies), and interchangeability with the originator product (Cuadrado et al., 2013; Lie et al., 2015; Mellstedt, 2013; Rompas et al., 2015; Weise et al., 2012), which is also underlined by a survey among physicians (N=307) with focus on inflammatory bowel diseases (Danese et al, 2014). These results are corroborated by selected expert interviews (N = 6) that were conducted in the course of this study. Most of the interviewees (N=5) have not yet heard of biosimilars. One physician (nephrologist) who had experience with epoetin, stated:
“Production, quality, immunogenicity and efficacy of biosimilars are not sufficiently transparent. Clinical studies are extrapolated by using the data of the reference product, which is not suitable.”
It is interesting to note that such skepticism remains although biosimilars have to run through a thorough authorization process as outlined in section 3.1.1, and that they ,to the best of our knowledge, did not yet show any adverse effects. However, in orderfor biosimilars to be successful on the market, companies also have to think about how to better inform physicians about the therapeutic advantages of biosimilars.
Patients, on the other hand, are nowadays better aware about their diseases and the therapeutic options. For chronological diseases such as cancer, patients seem to be rather skeptical about changing from an established original biologic to a biosimilar. They also think critical about the expression “biosimilar”, as the word “similar” implies not getting the same drug. For short-term therapies such as epoetin treatment, on the contrary, substitution of the original product with a biosimilar seems less problematic.
3.1.4 Technological
Concerning the technological perspective, the literature review reveals two majortopics. The first of them, the so-called quality-by-design (QbD) approach, is receiving increased attention as an approach to project management in biosimilar development and production. The QbD approach was first introduced by Juran (1992) and has been adopted by the U.S. Food and Drug Administration (FDA) for the development of pharmaceutical products (FDA, 2007).The QbD approach has also been explicitly discussed for biologics (Rathore, 2009; Scott, 2011). Essentially, the QbD approach aims at a thorough understanding as well as control ofthe potential influencing variables in the manufacturing process of a pharmaceutical drug from the very beginning. Thereby, the risks associated with the development process of a pharmaceutical drug, such as process changes (Schiestl, 2011) or variability of (biological) raw material quality (Rathore, 2009), should be better understood and thus already mitigated during the development phase, e.g. upon planning of the manufacturing process. The QbD approach is also reflected by the proverb “the process is the product” that is often coined in the context of biosimilars. It gives companies a higher flexibility in designing a manufacturing process for biosimilars and reflects the inherent complexity of biomolecules on a project management level as it provides companies with the flexibility to decide how to best obtain the end product. On the other hand, this flexibility may come at the cost of higher uncertainty for companies as a large number of process parameters have to be taken into account from the very beginning of a biosimilar development project.
The second important topic is the question of how to proof similarity of a biosimilar to the reference product. Again, given the large molecular size and the inherent complexity of biologics, it is obvious that a large array of state-of-the-art bioanalytical tools is required to address this question. Due to the progress in analytical science, there are now many different analytical methods available to analyze biosimilars (Kálmán-Szerekes et al.,2012; Tsuruta et al., 2015). Especially mass spectrometry has proven as an invaluable tool to analyze structural similarity, as has been highlighted e.g. by studies of epoetin biosimilars (Harazono et al., 2013) as well as complex monoclonal antibodies (Beck et al., 2013). Even batch-to-batch structural changes or structural alterations upon variation of the manufacturing process can be monitored (Kálmán-Szerekes et al., 2012). From the perspective of innovation management, it is interesting to note that the progress of the (bio)analytical science seems to be a necessary prerequisite in order to capture the market potential of biosimilars, which is in line with the assumption of some experts that analytical science is a key driver of innovation in the chemical sciences (Franz, 2013).
3.1.5 Summary of PEST analysis
The summary ofthe PEST analysis ofthe biosimilars market is shown in figure 4.
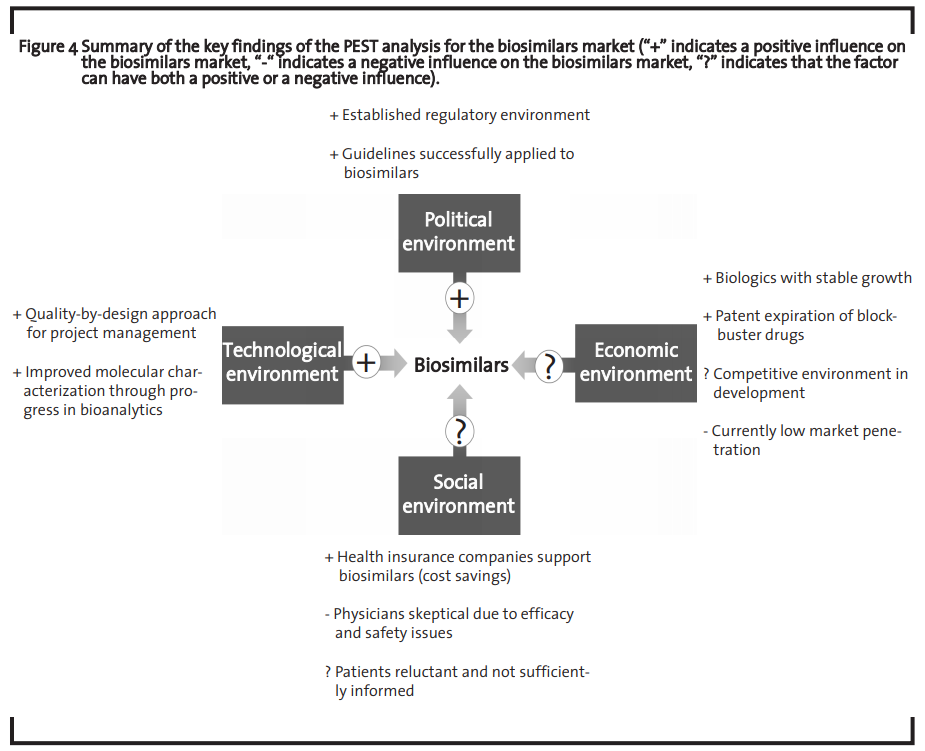
The analysis shows that the market is economically attractive and that both the technological means to address this market as well as the regulatory environment that ensures a framework in which companies can operate to capture this market are at hand. However, the regulatory environment must be flexible enough to adapt to technological progress (Huzair and Kale, 2015). The challenges to successfully develop a biosimilar may still be high but the huge market opportunity seems to make investments worthwhile, which is in line with previous findings (Blackstone and Fuhr, 2013). The current major bottleneck for the biosimilar market development seems to be an overall sceptical attitude of physicians as they do not feel sufficiently informed about biosimilars and are rather reluctant to prescribe those drugs.
3.2 Market outlook
Due to the factors discussed in the previous section, the biosimilars market does currently not capture its full market potential. However, chemically-synthesized generics also faced a similar situation 20 years ago. In 1990, only about 30% of the potential generics market were actually served by generics (the remaining 70% were occupied by originator drugs) (BPI, 2000; PhRMA, 2015). Interviews both with a market expert of the association ProGenerika e.V. and a leading German health insurance company corroborated this finding. According to these interviews, the current market situation of biosimilars can be interpreted as “starting difficulties” of an innovative market. However, it is not clear whether the biosimilar market will take a similar development as the market for chemically-synthesized generics. Given the complexity of the product and the lower number of competitors, no final conclusion concerning the comparability of these two markets can be drawn so far. In order to better understand the perspective of different stakeholders on biosimilars in recent years, 234 peer-reviewed scientific articles and conference contributions were analyzed. The analysis revealed that the perspective of the scientific community towards biosimilars changed from a rather sceptical evaluation 10 years ago to a rather positive assessment in 2015 (figure 5).
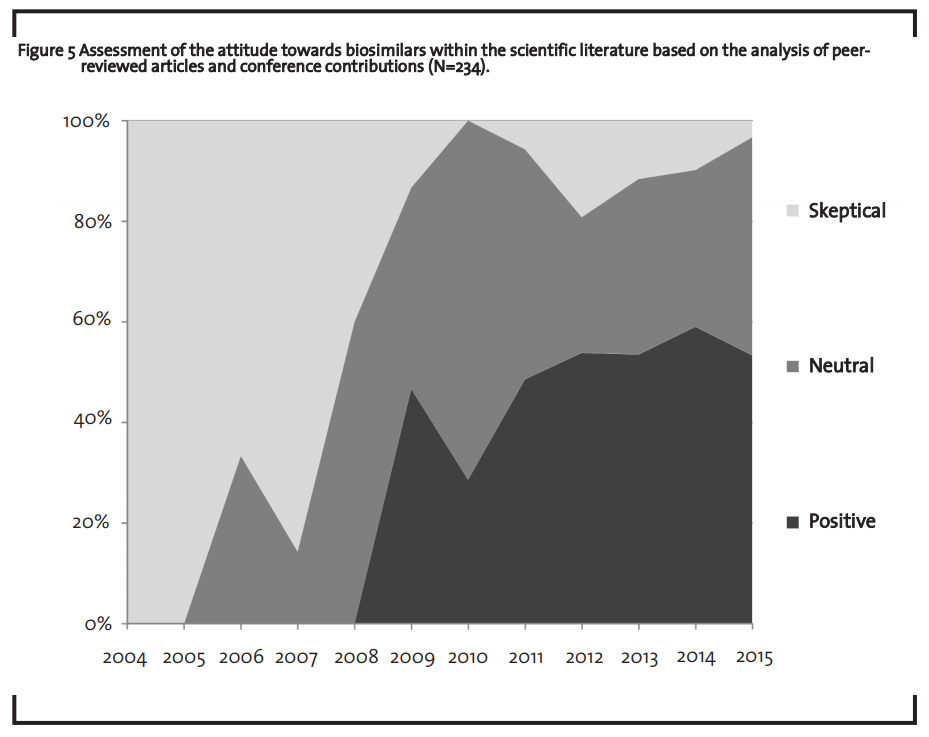
In the same period, an increasing number of countries developed guidelines on biosimilars (Heinemann et al., 2015) and significant progress in bioanalytics was made. In 2007, testing for similarity and comparability was still seen as “causes of concern” (Roger and Mikhael, 2007), whereas eight years later, Tsuruta et al. (2015) argue that “…improved analytical methods […] allow for the detection of even small changes […] between lots of the reference products currently on the market” (Tsuruta et al., 2015). Finally, another important aspect when discussing the market outlook for biosimilars, is the demographic change in developed countries. Facing a rising life expectancy and a growing average age, access to high quality medicine without compromising the benefits of national healthcare systems already is and will become a major issue for healthcare politics. Biosimilars can play a crucial role in this context.
4 Concluding remarks
Biosimilars have successfully entered the healthcare market and broadened the access to affordable high quality medicinal products. An established regulatory environment secures the framework for companies seeking to invest in this dynamic market. The current sceptical attitude of physicians and patients should be addressed by better informing these stakeholder groups about biosimilars, e.g. by teaching medical students at universities about biosimilars in order to improve the attitude of physicians or by information brochures about biosimilars in order to improve the attitude of patients. Based on the comparison with chemically-synthesized generics and the changed attitude of the scientific world towards biosimilars, the market share of biosimilars in the market for biologics that already lost patent protection is expected to increase in future. The market is thus attractive for companies thinking about to enter this innovative environment.
The implication for the management depends on the type of company. For traditional generic companies, development of a second-wave biosimilar does not seem to be attractive, given the high upfront investments and the high risk. For them, seeking alliances with larger biosimilar companies would probably yield more success, e.g. if they focus on distribution, their core competency, and in-license the biosimilar from a company that focuses on biosimilar development and does not have market access. Potential partners could be, e.g., market entrants from foreign markets.
For big pharmaceutical companies, biosimilars seem to be an attractive option to fill their pipeline and to leverage their development expertise as well as their financial strength. As development of a biosimilar requires much of the expertise of new drug development, big pharma companies should be in a good starting position to address this market.
For new market entrants, the hurdles are high: regulatory know-how, expertise in quality-by-design project management and financial strength. However, as the case of Celltrion shows, given the right strategy and financial support, also those companies can enter the market.
The biosimilars market will probably develop towards an oligopolistic market, as also found in other studies (Declerck and Simoens, 2012; Rompas et al., 2015). The competitive environment will be shaped by a mix of non-classical competitors such as big pharmaceutical companies and new market entrants from foreign markets as well as a small number of classical generic players in the first-wave biosimilar market. Product competition in this market will rather occur between the biosimilar and the originator drug than between different biosimilar versions of the drug as can be observed for chemically-synthesized generics, which has also been described by Carlson (2009). But even if the competitive environment will be characterized by a lower number of players compared to the chemically-synthesized generics market, the financial burden on the healthcare system will nonetheless be reduced, which will be a major driver of the biosimilars market in the long run. Biosimilars will thus continue to become an integral part of the pharmaceutical portfolio in the future. The question will no longer be “if” but rather “when” this dynamic market will see its breakthrough.
References
Apotheke adhoc (2015): Biosimilars zu Remicade, available at http://www.apotheke-adhoc.de/nachrichten/nachricht-detail/biosimilars-hospira-inflectra-fuer-remicade/?L=0%3Ft%3Ft%3 D1%3Ft%3Ft%3Ft%3D1?t=1, accessed 3 January 2016.
Beck,A., Diemer,H., Ayoub,D., Debaene, F., Wagner Rousset, E., Carapito, C., Van Dorsselaer, A., and Sanglier-Cianférani, S. (2013): Analytical characterization of biosimilar antibodies and Fc-fusion proteins, Trends in Analytical Chemistry, 48, pp. 81-95.
BCG and vfa bio (2006): Zur Lage der Biotechnologie im Bereich Medizin in Deutschland.
BCGand vfa bio (2007-2014): Medizinische Biotechnologie in Deutschland.
BCG and vfa bio (2015): Medizinische Biotechnologie in Deutschland 2005–2015–2025.
Biosimilar News (2012): Teva halts the Phase III studies of biosimilar rituximab, available at http://www.biosimilarnews.com/teva-haltsthe-phase-iii-studies-of-biosimilar-rituximab, accessed 3 January 2016.
Blackstone, E. A., and Fuhr, J. P. (2013): The Economics of Biosimilars, American Health & Drug Benefits, 6 (8), pp. 469-478.
BPI (Bundesverband der Pharmazeutischen Industrie) (2000): Pharma Daten 2000, available at http://www.bpi.de/fileadmin/media/bpi/Downloads/Internet/Publikationen/PharmaDaten/ Pharmadaten_2000_DE.pdf,accessed 3 January 2016.
Bui, L. A., Hurst, S., Finch, G. L., Ingram, B., Jacobs, I. A., Kirchhoff, C. F., Ng, C.K., and Ryan, A.M. (2015): Key considerations in the preclinical development of biosimilars, Drug Discovery Today, 20 (S1), pp. 3-15.
Carlson, B. (2009): Biosimilar Market Fails to Meet Projections, BioMarket Trends, 29 (17), available at http://www.genengnews.com/gen-articles/biosimilar-market-fails-to-meet-projections/ 3050/, accessed 3 January 2016.
Casey, D. (2015): Key strategic factors for stakeholders in the current global biosimilars market, Drug Discovery Today, in press.
Cuadrado, M. J., Sciascia, S., Bosch, X., Khamashta, M.A., and Ramos-Casals,M.(2013): Is it time for biosimilars in autoimmune diseases?, Autoimmunity Reviews, 12 (10), pp. 954-957.
Danese, S., Fiorino,G., and Michetti, P.(2014):Viewpoint: Knowledge and viewpoints on biosimilar monoclonal antibodies among members of the European Crohn’s and Colitis Organization, Journal of Crohn’s and Colitis, 8, pp. 1548-1550.
de Mora, F. (2015): Biosimilar: what it is not, British Journal of Clinical Pharmacology, 80 (5), pp. 949- 956.
Declerck, P. J., Simoens, S. (2012): A European perspective on the market accessibility of biosimilars, Biosimilars, 2, pp. 33-40.
Dingermann, T., and Zündorf, I. (2014): Biosimilars: nicht gleich, aber ähnlich. Nachahmerprodukte von Biologika sind keine Generika, Ars Medici, 2/2014, pp. 104-107.
EMA (2005): Guideline on similar biological medicinal products, CHMP/437/04, October 30, 2005.
FDA (2007): Pharmaceutical Quality for the 21st Century A Risk-Based Approach Progress Report, available at http://www.fda.gov/aboutfda/centersoffices/officeofmedicalproductsandtobacco/ cder/ucm128080.htm, accessed 7 January 2016.
Franz, K. D. (2013): Nicht nur Kosten, sondern auch Wert, Nachrichten aus der Chemie, 61 (9), pp. 896-897.
GaBiOnline (2013): EC approves first monoclonal antibody biosimilar, available at http://www. gabionline.net/Biosimilars/News/EC-approvesfirst-monoclonal-antibody-biosimilar, accessed 3 January 2016.
GaBiOnline (2015a): Extrapolation of indications in biosimilars: infliximab, available at http://gabionline.net/Biosimilars/Research/Extrapolation-of-indications-in-biosimilars-infliximab, accessed 3 January 2016.
GaBiOnline (2015b): Huge discount on biosimilar infliximab in Norway, available at http://www. gabionline.net/Biosimilars/General/Huge-discount-on-biosimilar-infliximab-in-Norway, accessed 3 January 2016.
Harazono, A., Hashii, N., Kuribayashi, R., Nakazawa, S., Kawasaki, N. (2013): Mass spectrometric glycoform profiling of the innovator and biosimilar erythropoietin and darbepoetin by LC/ESIMS, Journal of Pharmaceutical and Biomedical Analysis, 83, pp. 65-74.
Heinemann, L., Khatami, H., McKinnon, R., and Home, P. (2015): An Overview of Current Regulatory Requirements for Approval of Biosimilar Insulins, Diabetes Technology & Therapeutics,17 (7), pp. 510-526.
Hinderer, W. (2012): Biosimilar Drugs, in: Kayser, O., Warzecha, H.(eds.), Pharmaceutical Biotechnology – Drug Discovery and Clinical Applications, Wiley-VCH, Weinheim, pp. 305ff.
Huzair, F., and Kale, D. (2015): Biosimilars and the long game, Trends in Biotechnology, 33 (5), pp. 250-252.
Juran, J. M. (1992): Juran on Quality by Design: The New Steps for Planning Quality into Goods and Services, FreePress, New York.
Kálmán-Szerekes, Z., Olajos, M., and Ganzler, K.(2012): Analytical aspects of biosimilarity issues of protein drugs, Journal of Pharmaceutical and Biomedical Analysis, 69, pp. 185-195.
KV Bremen (2015): Vereinbarung zur Sicherstellung der Arzneimittelversorgung im Jahr 2015, available at http://www.kvhb.de/sites/default/files/ arzneimittelvereinbarung-2015.pdf, accessed 3 January 2016.
Ledford, H. (2010): ‘Biosimilar’ drugs poised to penetrate market, Nature, 468, pp. 18-19.
Lie, G., Sciascia,S., and Cuadrado, M.J.(2015):Biosimilar vs biological agents in rheumatology: When are biosimilar agents similar enough?, International Immunopharmacology, 27, pp. 220-223.
Mellstedt, H. (2013): Clinical considerations for biosimilar antibodies, European Journal of Cancer Supplements, 11 (3), pp. 1-11.
PhRMA (2015): Biopharmaceuticals in Perspective, p. 56, available at http://www.phrma.org/sites/ default/files/pdf/chartpack-2015.pdf, accessed 3 January 2016.
ProGenerika e.V. (2014a): Biosimilars Handbuch, available at http://www.progenerika.de/wpcontent/uploads/2014/09/Biosimilars_EinHandbuch_Sept.-2014.pdf, accessed 3 January 2016.
ProGenerika e.V. (2014b): Biosimilars Faktenbuch, available at http://www.progenerika.de/wpcontent/uploads/2014/11/Biosimilars-Faktenbuch.pdf, accessed 13 January 2016.
ProGenerika e.V.(2015):Marktdaten AGPro Biosimilars 10/2015,available at http://probiosimilars .de/img_upload/2015/12/Marktdaten-Biosimilars_Oktober-2015_.pdf?ddl=1,accessed 3 January 2016.
Rathore, A.S.(2009):Roadmap for implementation of quality by design (QbD) for biotechnology products, Trends in Biotechnology, 27 (9), pp. 546-553.
Roger, S.D., andMikhail, A.(2007): Biosimilars: Opportunity or Cause for Concern?, Journal of Pharmacy & Pharmaceutical Sciences, 10(3), pp.405- 410.
Rompas, S., Goss, T.,Amanuel, S., Coutinho, V., Lai, Z., Antonini, P., and Murphy, M. F. (2015): Demonstrating Value for Biosimilars: A Conceptual Framework, American Health & Drug Benefits, 8 (3), pp. 129-139.
Schellekens, H. (2004): How similar do ‘biosimilars’ need to be?, Nature Biotechnology, 22, pp.1357- 1359.
Schellekens, H., Lietzan, E., Faccin, F., and Venema, J. (2015): Biosimilar monoclonal antibodies: the scientific basis for extrapolation., Expert Opinion on Biological Therapy, 15 (11), pp. 1633-1646.
Schiestl, M., Stangler, T., Torella, C., Cepeljnik, T., Toll, H., and Gau, R. (2011): Acceptable changes in quality attributes of glycosylated biopharmaceuticals, Nature Biotechnology, 29, pp. 310-312.
Schneider, C. K., and Kalinke, U. (2008): Toward biosimilar monoclonal antibodies, Nature Biotechnology, 26 (9), pp. 985-990.
Scott, C. (2011): Quality By Design and the New Process Validation Guidance, BioProcess International, May 2011, pp. 14-21.
Sheppard, A., and Di Biase, S.(2014):Biological/biotechnological and biosimilars market: the global outlook with special focus on Europe, Biosimilar Medicines: 12th EGA International Symposium, London, 3 April 2014.
Stada Unternehmensinformation (2011): STADA und Richter kooperieren bei der Entwicklung von zwei Biosimilarprodukten für zwei monoklonale Antikörper: Rituximab und Trastuzumab, available at https://www.stada.de/uploads/tx_news/07_STADA_und_Richter_kooperieren_b ei_der_Entwicklung_von_zwei_Biosimilarprodukten.pdf, accessed 3 January 2016.
Tsuruta, L.R., dos Santos, M. L., and Moro, A.M. (2015): Biosimilar Advancements: Moving on to the Future, Biotechnology Progress, 31 (5), pp. 1139- 1149.
VFA (2015): Zulassungen für gentechnisch hergestellte Arzneimittel, available at http:// www.vfa.de/de/arzneimittel-forschung/datenbanken-zu-arzneimitteln/amzulassungen-gentec.html, accessed 2 October 2015.
Weise, M., Bielsky, M. C., De Smet, K., Ehmann, F., Ekman, N., Giezen, T. J., Gravanis, I., Heim, H. K., Heinonen, E., Ho, K., Moreau, A., Narayanan, G., Kruse, N.A., Reichmann, G., Thorpe, R., van Aerts, L., Vleminckx, C., Wadhwa, M., and Schneider, C. K. (2012): Biosimilars: what clinicians should know, Blood, 120 (26), pp. 5111-5117.
Weise, M., Kurki, P., Wolff-Holz, E., Bielsky, M.C., and Schneider, C. K. (2014): Biosimilars: the science of extrapolation, Blood, 124 (22), pp. 3191-3196.